
4-1BB Receptor (CD137) Recombinant Protein, Human, 20ug
The 4-1BBR (TNFRSF9) is a receptor human recombinant protein consists of a single non-glycosylated polypeptide chain with 167 amino acids.
...

ACC1, Monoclonal Antibody, 100ug
This antibody binds ACC1 to the citrullinated triple-helical collagen type II (CII) epitope (position 359-369), the C1 epitope. The ACC1 antibo...
ACC4 (IgG1), Monoclonal Antibody, 100ug
This antibody is an anti-citrullinated protein antibody (ACPA) that, on Collagen alpha-1 (II) chain, binds specifically to citrullinated C1 epi...
ACC4 (IgG2b), Monoclonal Antibody, 100ug
This antibody is an anti-citrullinated protein antibody (ACPA) that on Collagen alpha-1 (II) chain binds specifically to citrullinated C1 epito...
ACPA (Negative Control), Monoclonal Antibody (Clone E4NG-Mutant), 100ug
This antibody is a negative control for the E4NG antibody (product number 1061006). It is the mutated version of the E4NG antibody, with identi...
ACPA, Monoclonal Antibody (Clone E4NG), 100ug
This antibody recognizes multiple citrullinated proteins/peptides, including CCP2, citrullinated collagen type 2 (COL2) peptides, citrullinated...
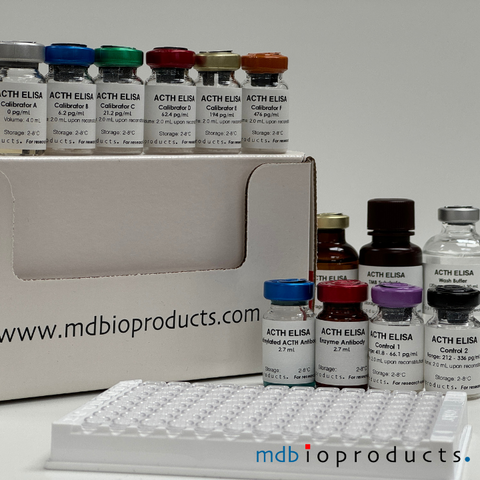
ACTH ELISA
ACTH ELISA kit to measure ACTH levels in human, mouse and rat plasma samples. ACTH (Adrenocorticotropic hormone) or corticotropin is a 39-amino aci...

Activin A Recombinant Protein (Human), 10ug
The human Activin A recombinant protein consists of a non-glycosylated polypeptide chain containing 137 amino acids (311-426aa).
Activin A, a m...

Aggrecan Antibody, C-terminal neoepitope NITEGE, 100 ug
Aggrecan monoclonal antibody to C-terminal neoepitope NITEGE (mouse, clone BC-13). This aggrecan degradation product usually remains within the tis...

Aggrecan Antibody, N-terminal neoepitope ARG, 100 ug
Aggrecan monoclonal antibody to N-terminal neoepitope ARG (mouse, clone BC-3). Aggrecan degradation products containing this neoepitope are rapidl...

Aggrecan Antibody, N-terminal neoepitope DIPEN, 100ug
Aggrecan monoclonal antibody to N-terminal neoepitope DIPEN (mouse, clone BC-4).
Proteoglycans are categorized depending upon the nature of their g...

Aggrecan Antibody, N-terminal neoepitope FFGV, 100 ug
Aggrecan monoclonal antibody to N-terminal neoepitope FFGV (mouse, clone BC14). This fragment is rapidly released from the tissue when MMP cataboli...

Aggrecan IGD Antibody, 100 ug
Aggrecan IGD monoclonal antibody (mouse, clone 6B4). This antibody detects aggrecan metabolites (intact or matrix protease-catabolised) in human sy...

Alpha Fetoprotein, Human, 0.1mg
Alpha Fetoprotein extracted from Human Blood Cord.
Alpha-fetoprotein (AFP) is a glycoprotein produced mainly by the fetal liver, yolk sac, an...
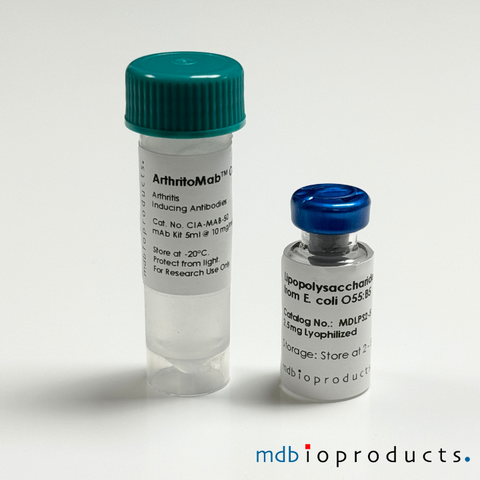
ArthritoMab™ Antibody Cocktail for Balb/c, DBA/1, R10.RIII, 50 mg
ArthritoMab™ Arthritis Inducing Antibody Cocktail is a cocktail of 4 arthritogenic monoclonal antibodies to collagen II (CII) used for inducing art...
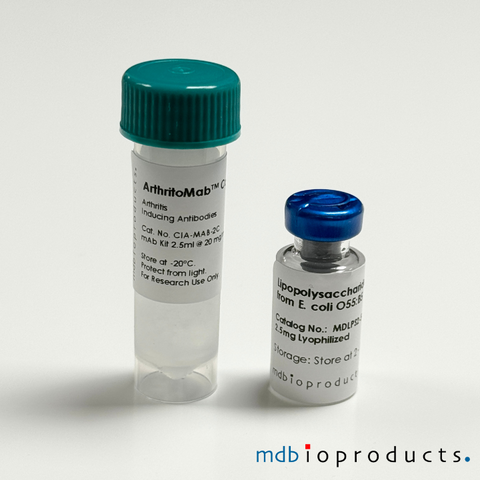
ArthritoMab™ Antibody Cocktail for C57BL/6, TG, 50 mg
ArthritoMab™ Antibody Cocktail is a reformulated cocktail of 4 monoclonal antibodies for the induction of arthritis as an alternative to the widely...

BDNF Recombinant Protein (Human), 10ug
The human BDNF recombinant protein consists of a single non-glycosylated polypeptide chain two 119 amino acids (and an N-terminal Met).
Brain-D...

BEND-6 Recombinant Protein (Human), 20ug
The human BEND-6 recombinant protein consists of a single, non-glycosylated polypeptide chain containing 302 amino acids (1-279 aa).
BEN domain...

Bovine Collagen Type VI, Lyophilized, 0.1mg
Bovine Collagen Type VI protein purified from bovine placenta.
Collagen Type VI is the major collagenous component of microfibrils in elastic fibe...
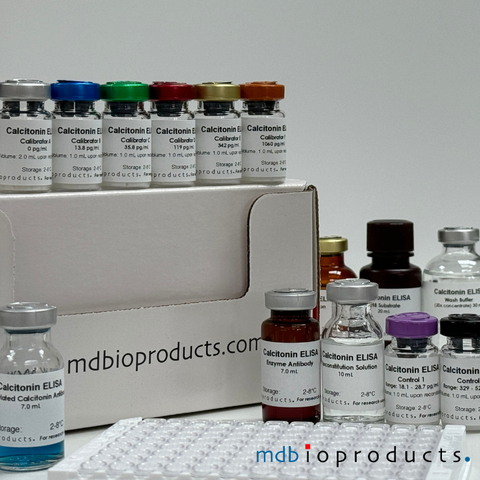
Calcitonin ELISA
Calcitonin ELISA kit to measure calcitonin levels in human samples within 4.5 hours.
Calcitonin, a 32-amino-acid polypeptide, is secreted primari...

Cartilage Link Protein, Antibody, 100 ug
Cartilage-link protein (HAPLN1) monoclonal antibody (clone 8A4). Cartilage-link protein (LP) is a glycoprotein present in cartilage that stabilizes...
Cartilage Oligomeric Matrix Protein (COMP) , Monoclonal Antibody (Clone 15A11), 100ug
This antibody binds to the fourth EGF (Epithelial Growth Factor) -like domain of mouse COMP, comprising residues 232–252, called the P6 epitope...
Cartilage Oligomeric Matrix Protein (COMP), Monoclonal Antibody (Clone 16B5), 100ug
This antibody binds to the coiled-coil domain or pentamer COMP. It cross-reacts with mouse, rat and human.
Cartilage Oligomeric Matrix Protein ...
CD28 Recombinant Protein, Human, 10ug
The human T-cell-specific surface glycoprotein CD28 is recombinant protein consists of a single glycosylated polypeptide chain with 376 amino a...

CD80 (CTLA-4 / B7-1) Recombinant Protein, Human, 5ug
The human CD80 (CTLA-4 Counter-Receptor B7-1) is a recombinant protein consisting of a single glycosylated polypeptide chain with 216 amino acid...

CD86 (CTLA-4 / B7-2) Recombinant Protein, Human, 20ug
The human CD86 (CTLA-4 Counter-Receptor B7-2) is a recombinant protein consisting of a single glycosylated polypeptide chain with 223 amino acid...
Chondroitin Sulphate Neoepitope Antibody, 100ug
Chondroitin sulfate monoclonal antibody (mouse, clone 1B5) to detect the zero sulphated Chondroitin Sulphate stub neoepitope generated by chondroit...

Chondroitinase generated C-4-S & DS Antibody, 100ug
Chondroitin-4-sulfate (C-4-S) monoclonal antibody that's also specific to dermatan-sulfate (DS) neoepitope (mouse, clone 2B6).
Monoclonal antibod...

CNTF Recombinant Protein (Human), 20ug
The human CNTF recombinant protein consists of a single, non-glycosylated, polypeptide chain containing 199 amino acids.
Ciliary neurotrophic f...

Collagen Type I (Atelocollagen) Porcine, 30 mg
Highly Purified Porcine Collagen Type I from porcine tendon. Lyophilized. Reconstitute to a concentration of 1-5 mg/mL.
Type I collagen is the m...

Collagen Type I (Atelocollagen), Bovine, 30 mg
Purified Bovine Collagen Type I (Atelocollagen) from bovine tendons.
Type I collagen is the most abundant collagen and is found in connective ti...

Collagen Type I (Atelocollagen), Mouse, 1 mg
Purified Mouse Collagen Type I (Atelocollagen) from mouse tail tendon.
Type I collagen is the most abundant collagen and is found in connective tis...

Collagen Type I (Atelocollagen), Rat, 30 mg
Purified Rat Collagen Type I (Atelocollagen) from rat tail tendon.
Type I collagen is the most abundant collagen and is found in connective tissues...

Collagen Type I and III, Bovine, 10 mg
Purified type I & III bovine collagen from calf skin.
Type I collagen is the most abundant collagen and is found in connective tissues includin...

Collagen Type I and III, Canine, 10 mg
Purified type I & III canine collagen.
Type I collagen is the most abundant collagen and is found in connective tissues including tendon, ligam...

Collagen Type I and III, Mouse, 5 mg
Purified native collagen type I and III from tail tendons of the mouse, 5 mg.
Type I collagen is the most abundant collagen and is found in connec...

Collagen Type I and III, Porcine, 10 mg
Purified type I & III native porcine collagen from pig’s tissue, 10 mg. Highest quality collagen for research use.
Type I collagen is the mo...

Collagen Type I and III, Rat, 10 mg
Purified type I & III native rat collagen purified from rat tissue. Highest quality collagen for research use.
Type I collagen is the most a...

Collagen Type I Antibody, anti-Mouse, 100 uL
Collagen type I polyclonal antibody (rabbit anti-mouse) purified from rabbits injected with type I collagen that was extracted/purified from mouse ...

Collagen Type I Antibody, anti-Rat, 100 uL
Collagen type I polyclonal antibody (rabbit anti-rat) purified from rabbits injected with type I collagen that was extracted/purified from rat skin...

Collagen Type I, Bovine, 10 mg
Highly Purified type I native collagen from bovine skin. Lyophilized
Type I collagen is the most abundant collagen and is found in connective tis...

Collagen Type I, Goat, 10 mg
Highly Purified type I native goat collagen. Lyophilized. Reconstitute to 10 mg/mL
Type I collagen is the most abundant collagen and is found in ...

Collagen Type I, Mouse, 1 mg
Purified type I native mouse collagen from mouse tail tendon. Lyophilized. Reconstitute to yield 1-5 mg/mL collagen solution.
Type I collagen is...

Collagen Type II Antibody, anti-Human, 100 uL
Collagen Type II polyclonal antibody (rabbit anti-human) purified from rabbits injected with type II collagen that was extracted/purified from huma...

Collagen Type II, Bovine, Immunization Grade, Lyophilized, 10 mg
Immunization Grade Collagen type II (CII) protein, purified from fetal bovine articular cartilage, for the induction of arthritis in the Collagen-I...
Collagen Type II, Bovine, Immunization Grade, Soluble, 2 mg/mL
Immunization Grade Collagen type II (CII) protein, purified from fetal bovine articular cartilage, for the induction of arthritis in the Collagen-I...
Collagen Type II, Chicken, Lyophilized, 10 mg
Highest quality (>99%) Collagen type II (CII) protein, purified from chicken sternum, for the induction of arthritis in the Collagen-Induced Art...
Collagen Type II, Chicken, Soluble, 2 mg/mL
Highest quality (>99%) Collagen type II (CII) protein, purified from chicken sternum, for the induction of arthritis in the Collagen-Induced Art...
Collagen Type II, Monoclonal Antibody (Clone CIIC1), 100ug
This antibody binds specifically to the C1 epitope on triple helica collagen type 2. It cross-reacts with mouse, rat, chicken, baboon and horse...
Collagen Type II, Monoclonal Antibody (Clone M2139), 100ug
This antibody (clone 2139) recognizes the conformational J1 epitope on the triple-helical structure of the native Collagen Type II molecule. Th...