Featured Publication in Focus: Size-switchable polymer-based nanomedicines in the advanced therapy of rheumatoid arthritis
Apr 19, 2024
Authors: Alena Libánská , Eva Randárová, Svitlana Skoroplyas, Michael Bartoš,...


Stocking the most innovative labs around the world. Shop products below.






a flexible and affordable ELISA system

supplying the purest collagen type 2 for inducing arthritis in preclinical studies.
A rapid alternative to standard arthritis models.
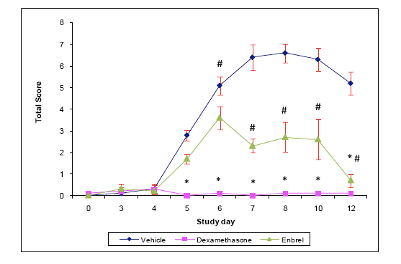

Apr 19, 2024
Authors: Alena Libánská , Eva Randárová, Svitlana Skoroplyas, Michael Bartoš,...

Apr 16, 2024
Authors: Sunil Tomar, PhD, Varsha Ganesan, MSc, Ankit Sharma, PhD, Lisa Waggo...
Mar 26, 2024
Authors: Yilin Wang, Piaopiao Pan, Aneesah Khan, Çağlar Çil and Miguel A. Pin...

